近日,清华大学统计学研究中心邓柯副教授课题组与美国弗吉尼亚大学臧充之教授团队合作,在生物统计学顶级期刊Nature Communications发表了题为Intrinsic bias estimation for improved analysis of bulk and single-cell chromatin accessibility profiles using SELMA的论文。该文章利用单纯形编码改进了高通量测序数据中序列偏倚的量化模型,可以更准确地估计并修正序列偏倚这一酶切内禀属性对开放染色质测序数据的影响。臧充之教授团队的胡圣恩博士为该文的第一作者,邓柯副教授和其课题组李祺博士为共同作者。

全基因组染色质开放区域的分析是研究表观遗传与基因转录调控的主要手段之一。染色质可及性(chromatin accessibility)高通量测序技术(包括基于DNaseI的DNase-seq技术以及基于Tn5转座酶的ATAC-seq技术)可以用来测定全基因组尺度的染色质开放区域图谱,并进而推断细胞核内的转录因子DNA结合位点以及基因表达调控的信息。虽然DNase-seq技术和ATAC-seq技术均为,但DNaseI和Tn5转座酶对于DNA的酶切作用仍然带有一定的序列偏好性,这种偏好性会混杂在高通量测序数据中,给数据分析带来潜在挑战。该现象曾经由哈佛大学刘小乐教授和Myles Brown教授团队在2013年提出。
将ATAC-seq技术与近年来被广泛应用的单细胞测序技术相结合,目前我们可以使用单细胞ATAC-seq(scATAC-seq)方法描绘出单细胞(single cell)或单细胞核(single nucleus)尺度上的染色质开放区域,因此可以极大的拓展数据量,但由于scATAC-seq数据在单细胞层面上极其稀疏,Tn5转座酶的序列偏好性可能造成更为严重的影响。如何对大量单细胞的开放染色质测序数据进行有效纠偏,提升高通量数据的生物学可解释性,仍是计算生物学领域内的一个重要问题。
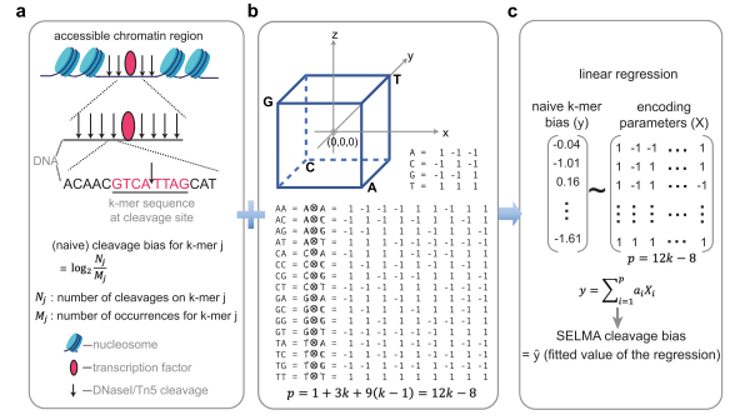
在该文章中,作者提出了名为SELMA (Simplex Encoded Linear Model for Accessible Chromatin)的开放染色质测序数据纠偏算法。在该算法中,作者使用单纯形编码(simplex encoding)模型取代了传统的k-mer模型,从而大大减小了模型参数,参数由缩减为12k-8。在此前提下,该文章可以回收传统DNase-seq/ATAC-seq数据分析中通常被丢弃的线粒体DNA测序片段,用这些数量较小、组成多样性较低的序列片段来准确估计样品数据中的偏倚水平,以此解决了传统方法需要外加DNA酶切样品数据集作为独立参考来进行偏倚水平估计的问题。与此同时,通过分析不同平台产生的单细胞scATAC-seq数据,该方法首次研究了酶切序列内禀偏倚对单细胞开放染色质测序的影响,使用针对单细胞数据的SELMA算法纠偏后,修正的scATAC-seq数据可以获得更加准确的细胞聚类结果。