
2021年4月,清华大学统计学研究中心17级博士研究生郭瀚民同学以第一作者身份在Nature Communications期刊上发表了题为Detecting local genetic correlations with scan statistics的研究论文,统计中心侯琳副教授和威斯康辛大学麦迪逊分校吕琼石助理教授为该论文的共同通讯作者,威斯康辛大学麦迪逊分校的James J. Li为本文的共同作者。针对复杂疾病的遗传相关性问题,研究人员提出了基于扫描统计量的统计推断方法,精准识别局部遗传相关性位点,并开发了相应软件包LOGODetect(https://github.com/ghm17/LOGODetect)。
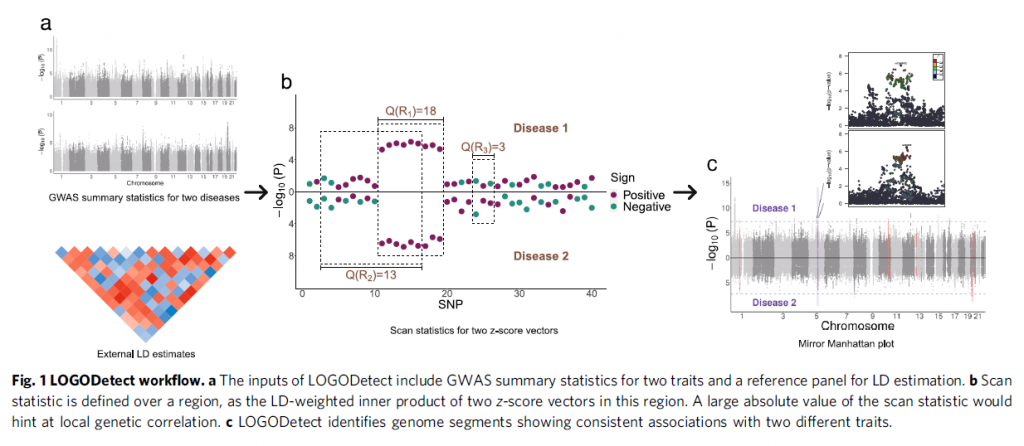
全基因组关联分析是研究人类复杂性状遗传机制的重要方法,被广泛应用于复杂疾病的研究,揭示了大量疾病易感位点和致病基因。研究数据表明,多种复杂疾病存在广泛的遗传相关性。已有的研究往往将遗传相关系数的估计转化为线性混合模型中协方差参数的估计问题,其前提假设是人类基因组中大量多态性位点对疾病遗传相关性的贡献服从同一分布。研究团队认为此假设是对疾病遗传结构的过度简化,无法准确刻画疾病间复杂的遗传相关性结构。针对此问题,研究团队提出富集型遗传模型,即两个疾病的遗传相关性仅分布在基因组中的部分片段上,并开发了基于扫描统计量的局部遗传相关性统计推断方法。该方法以全基因组关联分析的概括统计量作为输入数据,在全基因组中寻找显著富集遗传相关性的区域,从而精确识别疾病间局部遗传相关性。
与已有方法相比,LOGODetect的优势在于:(1)自动搜索局部相关性区域,灵活准确地确定区域边界;(2)严格控制一类错误,具有更高的统计功效;(3)该方法对不同的遗传模型以及不同疾病数据集中样本重叠的问题具有鲁棒性。研究团队将该方法应用于多种神经系统相关疾病和表型的全基因组关联分析数据,LOGODetect识别出了227个互不重叠的与多个表型相关的基因片段,对理解精神疾病中的跨诊断现象具有重要意义。